Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS)¶
Summary
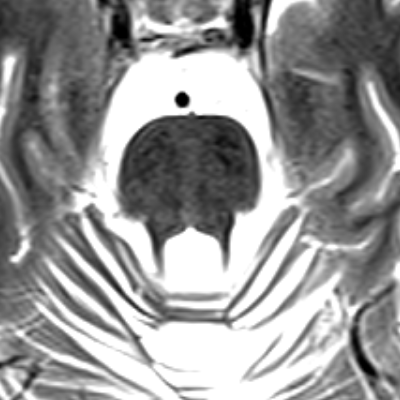
- ARSACS is a rare neurodegenerative disorder characterised by early-onset cerebellar ataxia, spasticity, and peripheral neuropathy
- Caused by mutations in the SACS gene, leading to progressive cerebellar and spinal cord degeneration
- Imaging reveals cerebellar atrophy, particularly of the superior vermis, and linear hypointensities in the pons on T2-weighted MRI1
Pathophysiology¶
- Autosomal recessive inheritance pattern
- Mutations in the SACS gene on chromosome 13q12.12
- SACS gene encodes sacsin protein, involved in protein folding and quality control
- Loss of sacsin function leads to:
- Mitochondrial dysfunction
- Accumulation of neurofilaments
- Progressive neurodegeneration, particularly in the cerebellum and spinal cord
Demographics¶
- Originally described in the Charlevoix-Saguenay region of Quebec, Canada
- Prevalence in this region: 1 in 1,932 individuals
- Worldwide prevalence: rare, with cases reported in various populations
- Typical age of onset: early childhood (12-18 months)
- Equal gender distribution
Diagnosis¶
- Clinical presentation:
- Early-onset ataxia (unsteady gait)
- Progressive spasticity
- Peripheral neuropathy
- Dysarthria
- Nystagmus
- Genetic testing:
- Identification of biallelic pathogenic variants in the SACS gene
- Electromyography and nerve conduction studies:
- Evidence of axonal-demyelinating sensorimotor neuropathy
Imaging¶
- MRI findings:
- Cerebellar atrophy, particularly of the superior vermis
- Linear hypointensities in the pons on T2-weighted images ("Tiger stripe" appearance)
- Cervical spinal cord atrophy
- Cerebral cortical atrophy (in advanced cases)
- Spinal cord imaging:
- Thinning of the cervical and thoracic spinal cord
- Optical Coherence Tomography (OCT):
- Thickening of the retinal nerve fibre layer
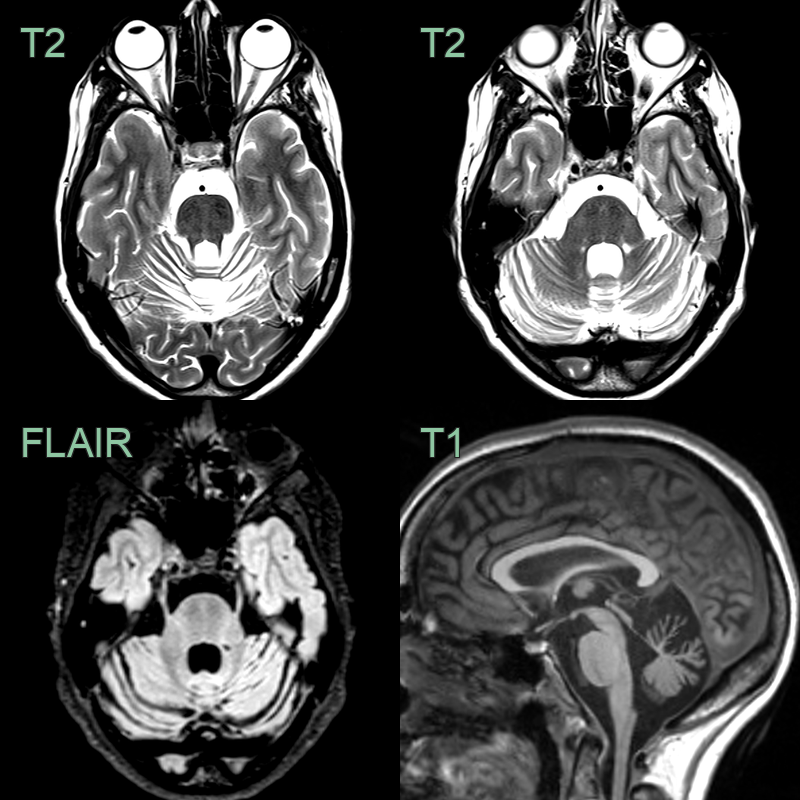
- Subtle symmetrical tigroid T2-hypointensity within the pons and severe superior cerebellar vermian atrophy.
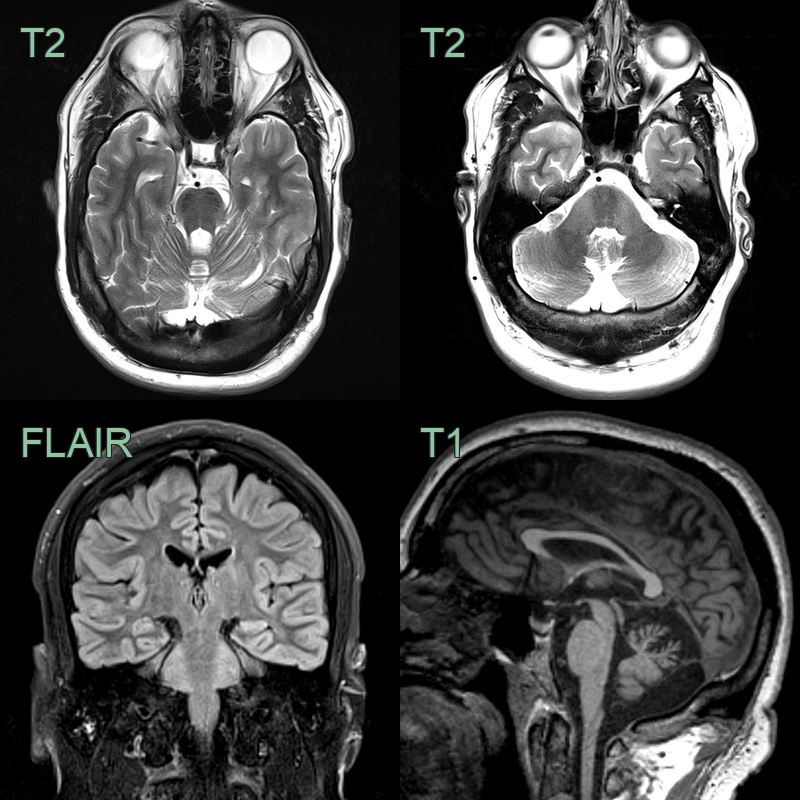
- 35-year-old patient presented with ataxia.
- MRI showed atrophy of the superior vermis, hyperintensity in the middle cerebellar peduncles and, most characteristically, tigroid T2-hypointensity within the pons.
Treatment¶
- No curative treatment; management is supportive (anti-spasticity agents, physiotherapy, orthoses)
- Genetic counselling for affected families
Differential diagnosis¶
| Imaging differential | Differentiating feature |
|---|---|
| Friedreich ataxia | Cervical cord and dorsal-column atrophy without superior vermian atrophy or pontine stripes |
| Multiple system atrophy, cerebellar type | "Hot cross bun" pontine T2 hyperintensity rather than linear T2 hypointensities |
| FXTAS | Symmetric middle cerebellar peduncle T2 hyperintensity without the pontine tigroid hypointensity |
| Spinocerebellar ataxia | Olivopontocerebellar atrophy without pontine tigroid hypointensity or superior vermian predominance |
-
Richter et al. Clinical and molecular genetic studies on autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). 1993. Advances in neurology - Open in new tab. ↩