Creutzfeldt-Jakob disease (CJD)¶
Summary
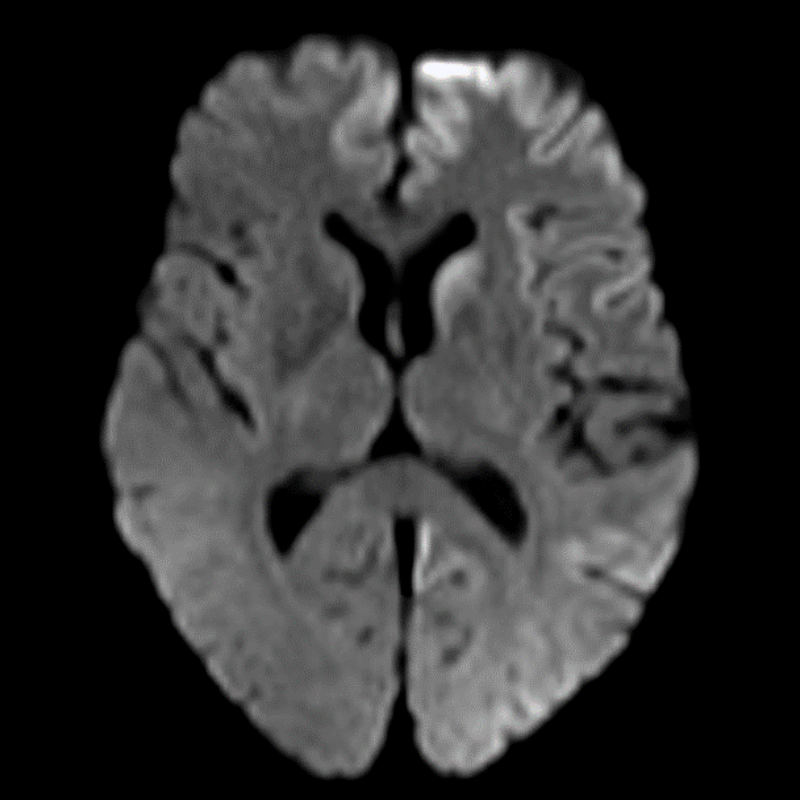
- Rare, fatal neurodegenerative disorder characterised by rapidly progressive dementia
- Caused by abnormal prion protein accumulation in the brain
- Imaging findings include cortical ribboning and basal ganglia hyperintensities on DWI/FLAIR1
Pathophysiology¶
- Accumulation of abnormally folded prion proteins (PrPSc) in the brain
- Leads to neuronal loss, gliosis, and spongiform changes
- Four subtypes:
- Sporadic (sCJD): Most common (85-90% of cases)
- Familial (fCJD): Genetic mutations in PRNP gene
- Iatrogenic (iCJD): Transmission via contaminated medical procedures
- Variant (vCJD): Linked to bovine spongiform encephalopathy (BSE)
Demographics¶
- Incidence: 1-2 cases per million population per year
- Median age of onset:
- sCJD: 60-65 years
- vCJD: 26-28 years
- No gender predilection
- Geographical distribution: Worldwide, with higher incidence in some countries due to genetic factors
Diagnosis¶
- Clinical presentation:
- Rapidly progressive dementia
- Myoclonus
- Visual disturbances
- Cerebellar ataxia
- Diagnostic criteria:
- WHO criteria for probable CJD
- MRI findings
- CSF biomarkers (14-3-3 protein, tau protein, RT-QuIC)
- EEG: Periodic sharp wave complexes
- Definitive diagnosis: Brain biopsy or post-mortem examination
Imaging¶
- MRI:
- DWI/FLAIR:
- Cortical ribboning (high signal in cortical grey matter)
- Basal ganglia hyperintensities (caudate and putamen)
- Thalamic hyperintensities (pulvinar sign in vCJD)
- T2-weighted:
- Subtle hyperintensities in affected areas
- CT:
- Generally normal in early stages
- May show cerebral atrophy in advanced cases
- PET:
- Hypometabolism in affected cortical regions and basal ganglia
- SPECT:
- Reduced perfusion in affected areas
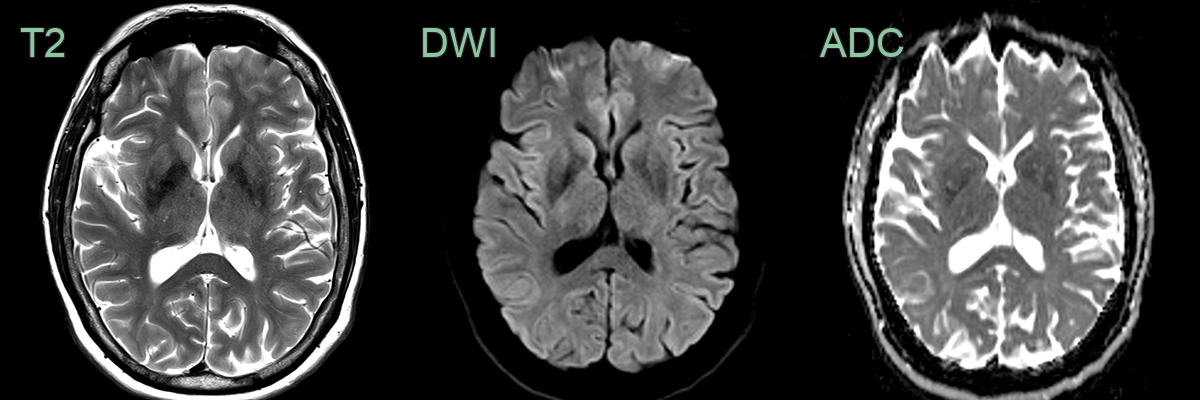
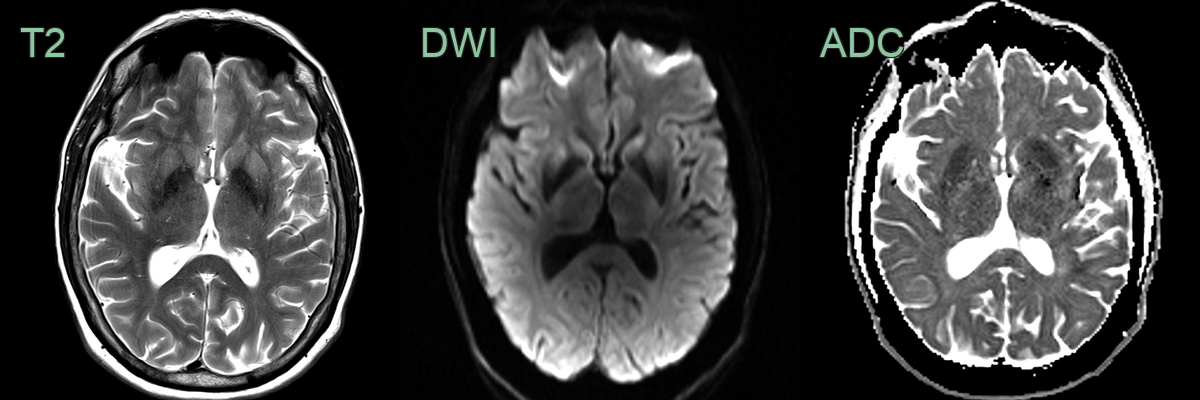
- 65-year-old male presented with myoclonus and cognitive impairment.
- On the initial MRI, rather equivocal DWI hyperintensity in the corpus striatum became obvious 6 months later.
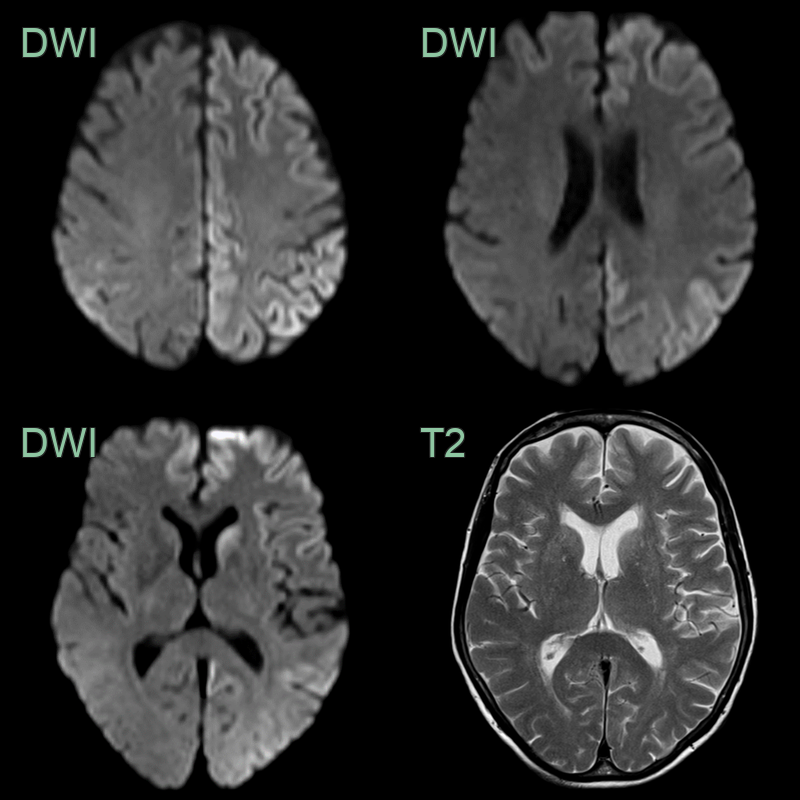
- A 71-year-old female developed progressively worsening confusion and visual hallucinations.
- Cortical ribboning on DWI was most apparent in the left caudate head, frontal lobe and parietal lobe.
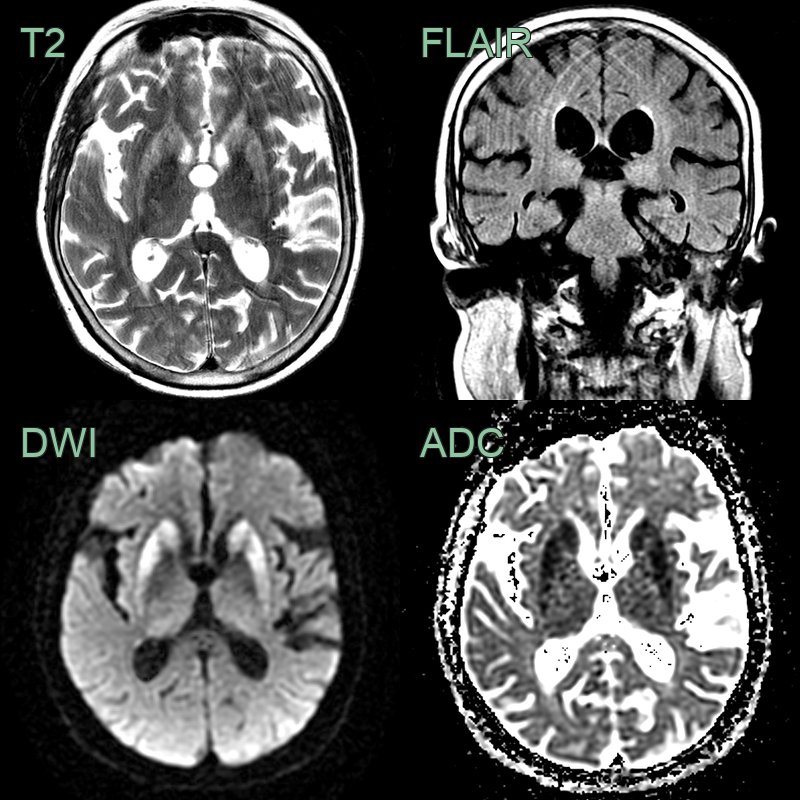
- A 65-year-old patient presented with rapidly progressive cognitive impairment and cerebellar ataxia.
- There was diffusion restriction in the striatum and FLAIR hyperintensity, without diffusion restriction, in the pulvinar of the thalami.
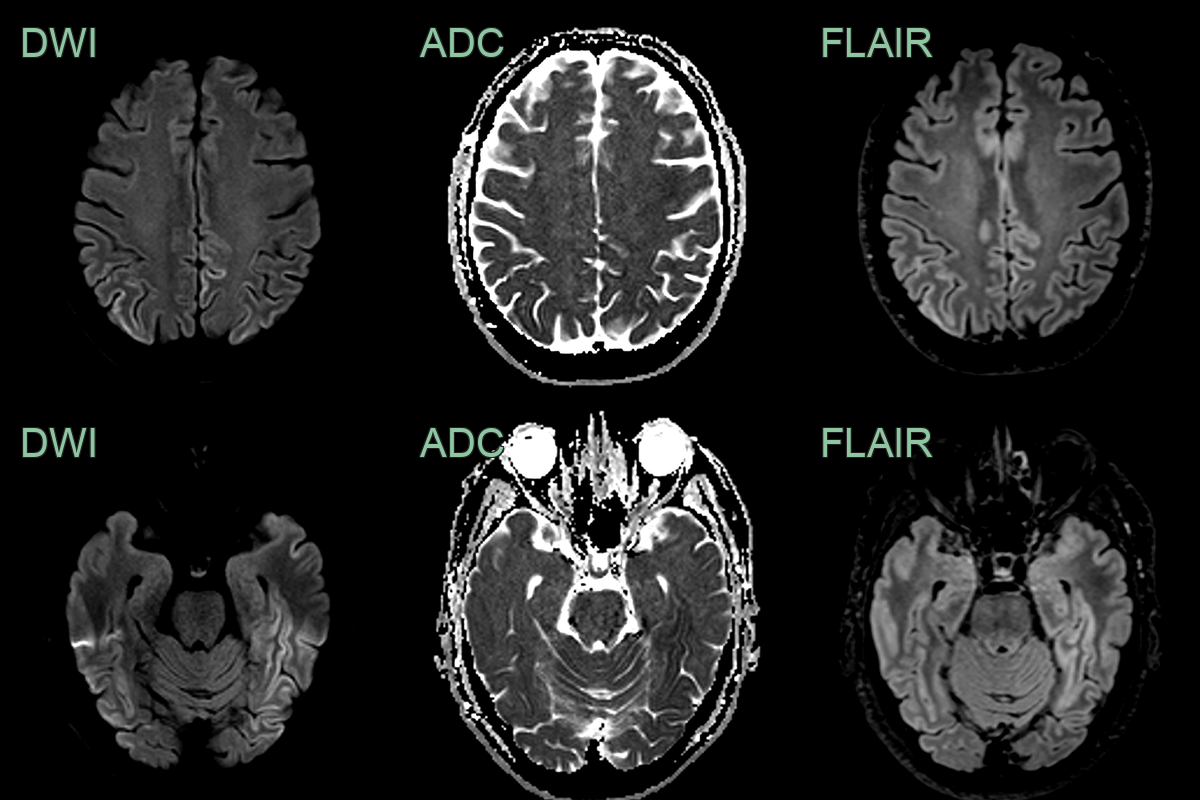
- Patient with recent onset cognitive impairment.
- MRI showed asymmetrical extensive cortical ribboning in the parietal, occipital and temporal lobes.
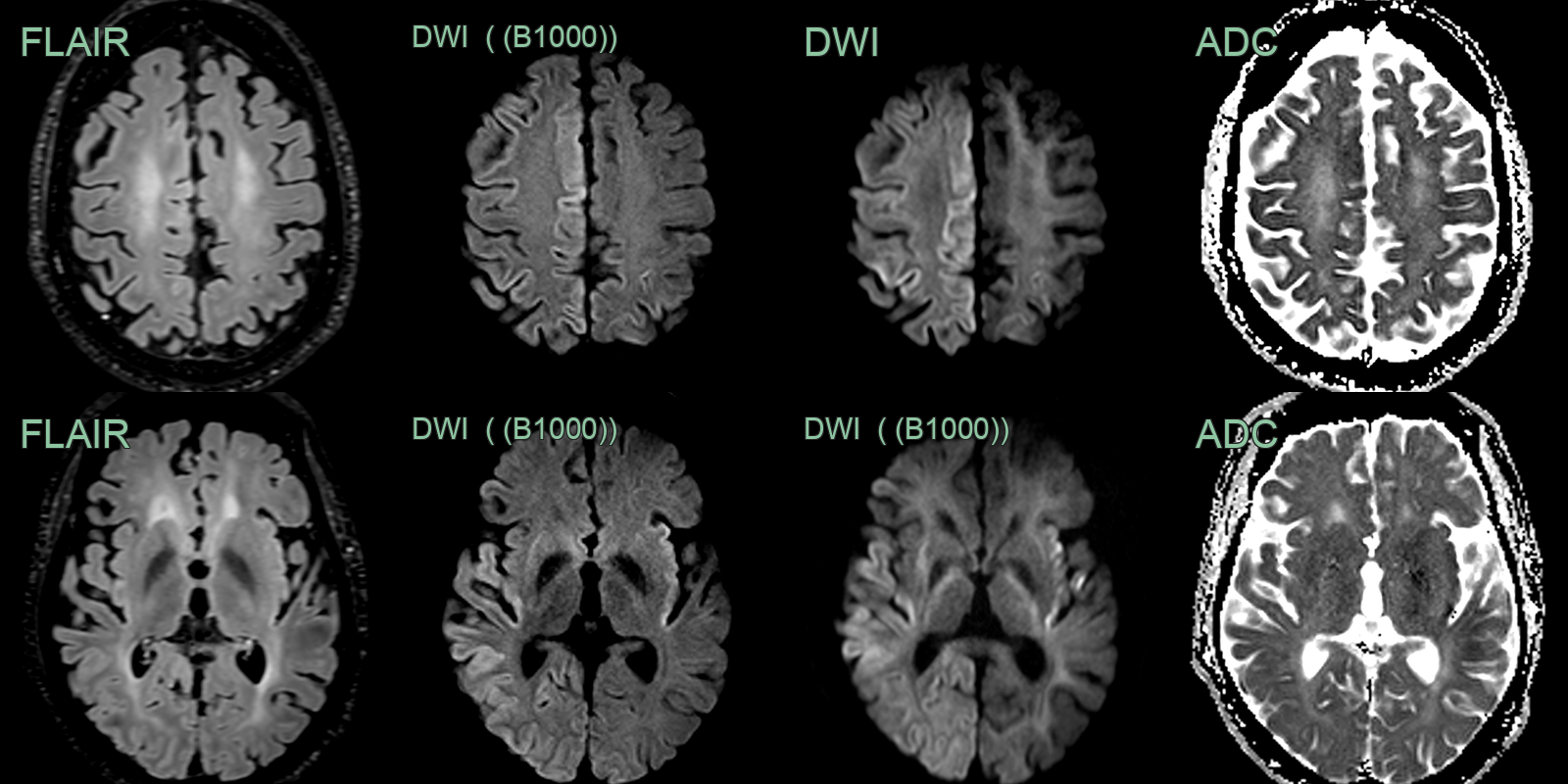
- A 60-year-old patient presented following a focal seizure followed by progressively worsening confusion.
- Imaging showed cortical (and subtle putaminal) diffusion restriction that was more apparent on B3000 than B1000 DWI.
- RT-QuIC confirmed the diagnosis of CJD.
Treatment¶
- No curative treatment; supportive care only. Uniformly fatal, usually within a year
Differential diagnosis¶
| Differential Diagnosis | Distinguishing Feature |
|---|---|
| Paraneoplastic/autoimmune limbic encephalitis | Bilateral mesial temporal FLAIR hyperintensity without cortical ribboning; may enhance |
| Viral encephalitis (HSV) | Asymmetric mesial temporal/insular haemorrhagic FLAIR hyperintensity without cortical ribboning |
| Wernicke's encephalopathy | Symmetric T2 hyperintensity of mammillary bodies, periaqueductal grey, and medial thalami |
| Autoimmune encephalitis | Bilateral hippocampal FLAIR hyperintensity; cortical DWI restriction absent |
-
Hermann et al. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. 2021. The Lancet. Neurology - Open in new tab. ↩